Benutzer-definierte Funktionen
Benutzerdefinierte Funktionen
- Funktionen sind Elemente der meisten Programmiersprachen.
- Funktionen reduzieren die Redundanz im Programmcode.
- Funktionen machen Programmblöcke wiederverwendbar und verfügbar.
- Funktionen kapseln Programmblöcke (Variablen haben Gültigkeitsbereiche, a innerhalb einer Funktion ≠ a außerhalb).
- Funktionen kommunizieren durch Argumente und Rückgabewerte.
Benutzerdefinierte Funktionen
Man erstellt eigene Funktionen mit der eingebauten Funktion function().
# Die neue Funktion funky() soll ein Argument annehmen
# und ein Ergebnis zurueckgeben.
> funky <- function(x){a <- x*2; print(a);
return(paste(a,'ist das Doppelte von', x)) }
> a <- 12 # ausserhalb!
> a
[1] 12
> r <- funky(10)
[1] 20 # Ausgabe des print-Befehls
> a # das 'aeussere' a ist unveraendert
[1] 12
> r
[1] "20 ist das Doppelte von 10" # r hat den Rueckgabewert aufgenommen
> x
Fehler: Objekt 'x' nicht gefunden # x existiert nur innerhalb von funky()
Benutzerdefinierte Funktionen
source() kann R-Skriptdateien nachladen. In R-Skripten kann man z. B. eine Bibliothek von nützlichen Funktionen sammeln, die dann immer nach dem Laden der Skriptdatei zur Verfügung stehen.
# Funktionssammlung in Datei meineFunktionen.R
dingens <- function(){
print("Jetzt bin ich da.")
}
> dingens()
Fehler in dingens() : konnte Funktion "dingens" nicht finden
> source('meineFunktionen.R')
> dingens()
[1] "Jetzt bin ich da."
Externe Skripte
Mit source('Ziel.R') lassen sich R-Skripte aus Dateien laden und ausführen. Das Ziel kann hierbei auch ein entfernter Server sein. Als Beispiel verwenden wir als Software-Repository "BioConductor".
# Um Pakete von Bioconductor zu installieren muss zuerst die Funktion
# biocLite() verfuegbar sein. Ein Skript, das diese definiert ist auf
# bioconductor.org vorhanden.
> source("http://bioconductor.org/biocLite.R")
Bioconductor version 3.4 (BiocInstaller 1.24.0), ?biocLite for help
# Jetzt lassen sich Pakete von bioconductor installieren:
> biocLite("Biostrings")
...
> library (Biostrings)
Lade noetiges Paket: BiocGenerics
Lade noetiges Paket: parallel
Attache Paket: 'BiocGenerics'
...
BioConductor heute:
Seit R 3.5:
> install.packages("BiocManager")
# und dann:
BiocManager::install(c('Paket1','Paket2'))
Bioinformatik mit R
Bioinformatik mit R
Es gibt zahlreiche R-Pakete, die Bioinformatik-Funktionen jeder Art zur Verfügung stellen.
Vieles ist auf CRAN zu finden, spezialisiert auf Bioinformatik ist jedoch das Package repository "BioConductor".
Sequenzdatenbanken
Um mit R auf Sequenzen aus den Primärdatenbanken (u.a.) zugreifen zu können verwenden wir das Paket "seqinr":
> library(seqinr)
> choosebank()
[1] "genbank" "embl" "emblwgs" "swissprot" ...
[8] "hogenomdna" "hovergendna" "hovergen" "hogenom5" ...
[15] "homolens" "homolensdna" "hobacnucl" "hobacprot" ...
[22] "greviews" "bacterial" "archaeal" "protozoan" ...
[29] "ensplants" "ensemblbacteria" "mito" "polymorphix" ...
[36] "taxodb"
> choosebank("genbank")
FASTA-Sequenzen
# Genbank anwaehlen
> choosebank('genbank')
# Sequenzen nach Spezies suchen
> res <- query('aras', "AC=DQ060130")
# erste Sequenz des Ergebnisses speichern
> seq <- getSequence(res$req[[1]])
> seq
[1] "g" "a" "g" "t" "t" "a" "g" "t" "t" "g" "a" "t" "c" "a" "a" "a" "t"
"g" "g" "t" "a" "g" "a" "a" "g" "t" "g" "g" "g" "a" "c" "a" "t"
> fasta <- paste('>A.alpina Sequenz', paste(seq, collapse=""),
collapse="", sep= "\n")
> fasta
[1] ">A.alpina Sequenz\ngagttagttgatcaaatg...
> cat(fasta)
>A.alpina Sequenz
gagttagttgatcaaatggtagaagtgggaca...
Nach dem Herunterladen liegt die Sequenz als Vektor von Zeichen vor. Um die übliche Textrepräsentation auszugeben, müssen wir einen Ein-Element-Character-Vektor daraus machen.
paste() hat das optionale Argument collapse, das die Elemente mit einem bestimmten Zeichen (oder mehreren) "zusammenklebt".
FASTA-Sequenzen
Oder so… ;-)
> seq <- getSequence(res$req[[1]], as.string=T)
Doppelklammern
Doppelte eckige Klammern kann man verwenden um einzelne Elemente zu addressieren:
> iris[['Sepal.Length']]
[1] 5.1 4.9 4.7 4.6 5.0 5.4 ...
> iris[['Sepal.Length']][1]
[1] 5.1
> head(iris['Sepal.Length'], 3)
Sepal.Length
1 5.1
2 4.9
3 4.7
a <- list('a', 'b', 'c')
> is.vector(a[1])
[1] TRUE
> is.list(a[1])
[1] TRUE
> is.vector(a[[1]])
[1] TRUE
> is.list(a[[1]])
[1] FALSE
Sequenzdatenbanken
Als weiteres Beispiel laden wir uns nun alle Sequenzen aus Brassicaceen herunter, für die das Schlüsselwort "APX" gesetzt ist. ("Ascorbatperoxidase").
# Wir brauchen das Paket 'ape', um mit Sequenzen arbeiten zu koennen
> library(ape)
# Erstellen einer Sequenzliste in 'sq' unter dem Namen 'sq1'
> sq <- query('sq1','SP=Brassicaceae AND K=apx')
> sq
8 SQ for SP=Brassicaceae AND K=apx
# Auslesen der Accession numbers aus sq
> ac <- getName(sq)
> ac
[1] "" "AB901369" "AB901370" "AB901371" "GQ500125" "HQ871864" "X78452" "X98003"
# Download von GenBank
> sqgb <- read.GenBank(ac[2:8], as.character=T)
# Auslesen der Speziesnamen aus sqgb
> sp <- attr(sqgb, "species")
> sp
[1] "Brassica_rapa_subsp._pekinensis" "Brassica_rapa_subsp._pekinensis"
[4] "Brassica_rapa_subsp._pekinensis" "Brassica_oleracea_var._italica"
[7] "Arabidopsis_thaliana"
Sequenzdatenbanken
Um eine FASTA-Datei zu erzeugen verwenden wir die Funktion write.fasta().
# Namen fuer die Sequenzen, bestehend aus Spezies und Accession number
> nm <- paste(sp, names(sqgb), sep='_')
> nm
[1] "Brassica_rapa_subsp._pekinensis_AB901369" "Brassica_rapa_subsp._pekinensis_AB901370" "Brassica_rapa_subsp._pekinensis_AB901371"
[4] "Brassica_rapa_subsp._pekinensis_GQ500125" "Brassica_oleracea_var._italica_HQ871864" "Raphanus_sativus_X78452"
[7] "Arabidopsis_thaliana_X98003"
# FASTA-Datei erzeugen
> write.fasta(sequences=sqgb,names=nm,file.out = "brass_apx.fa")
# dir() zeigt die vorhandenen Dateinamen
# read.fasta() liest die Sequenzen wieder ein
> read.fasta(file="brass_apx.fa")
> fa[7]
$Arabidopsis_thaliana_X98003
[1] "c" "a" "c" "a" "c" "a" "t" "c" "c" "t" "t" "g" "a" "c" "c" ...
[33] "t" "c" "t" "c" "t" "t" "t" "c" "t" "t" "c" "t" "t" "c" "a" ...
...
Sequenzanalyse-Basics
> length(seq)
[1] 510
> GC(seq)
[1] 0.4098039
> count(seq, 1)
a c g t
164 87 122 137
> count(seq, 3)
aaa aac aag aat aca acc acg act aga agc agg agt ata atc atg att caa cac cag cat cca ccc ccg cct cga cgc cgg cgt cta ctc ctg ctt gaa gac
18 8 17 18 8 6 7 5 12 3 4 12 9 10 18 8 20 6 4 7 4 6 2 4 6 2 4 1 1 8 5 7 14 8
gag gat gca gcc gcg gct gga ggc ggg ggt gta gtc gtg gtt taa tac tag tat tca tcc tcg tct tga tgc tgg tgt tta ttc ttg ttt
4 14 7 1 2 7 12 5 8 8 8 6 9 8 9 4 7 6 18 3 2 5 9 7 17 10 8 4 11 17
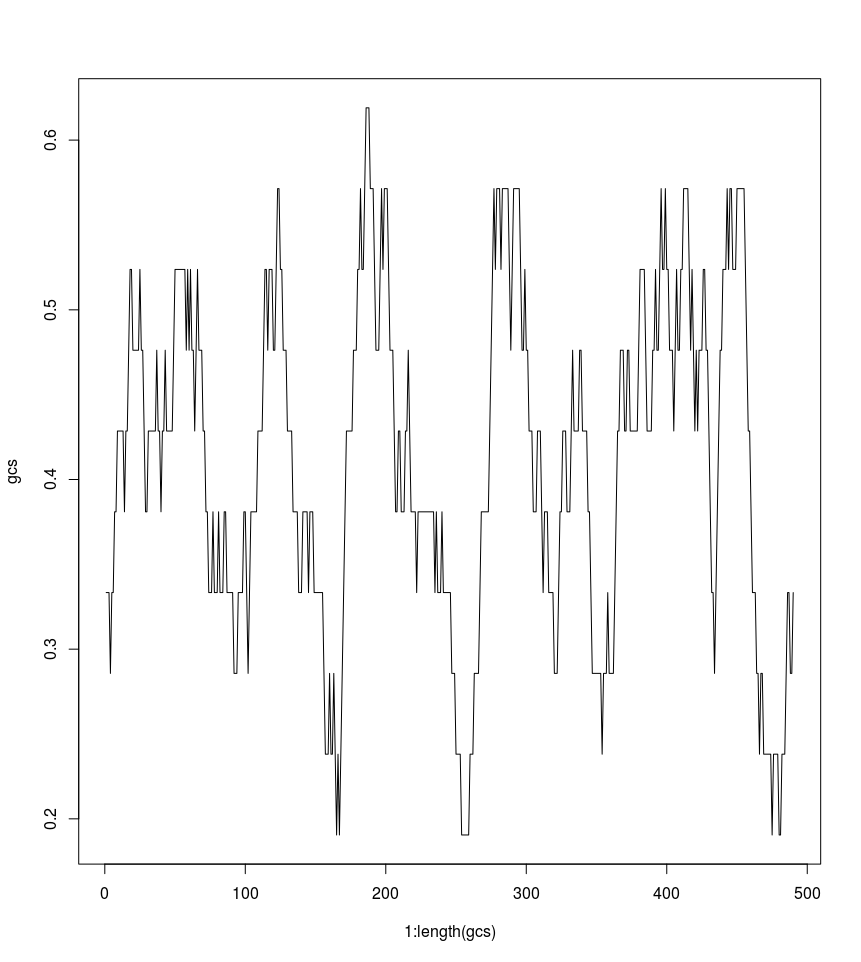
Sliding Window
> gcwin <- function(seq, win){
n<-length(seq); for(i in 1:(n-win)){
gc<-GC(seq[i:(i+win)]);print (gc);}
}
> gcwin(seq, 50)
[1] 0.3636364
[1] 0.3636364
[1] 0.3636364
[1] 0.4545455...
Sliding Window
> gcwin <- function(seq, win){
+ n<-length(seq)
+ out<-0
+ for(i in 1:(n-win)){
+ gc<-GC(seq[i:(i+win)])
+ out[i]<-gc
+ };return(out)
+ }
> gcs<-gcwin(seq,20)
> gcs
[1] 0.333333 0.333333 0.285714 ...
> plot(1:length(gcs),gcs,type='l')
Alignments
Wir können das Paket `muscle' verwenden, um unsere Sequenzen zu alignieren.
MUSCLE ist ein Multi-Alignmentprogramm.
# biocLite('muscle') # ggf. installieren
> library(muscle) # laden
# FASTA-Dateien als DNAStringSet laden
> fa <- readDNAStringSet("br_apx.fa")
> al <- muscle(fa) # al ist ein Alignment-Objekt
> al2 <- as.DNAbin(al) # al2 ist DNAbin-Format
7 DNA sequences in binary format stored in a matrix.
All sequences of same length: 1159
Labels:
Brassica_rapa_subsp._pekinensis_AB901369
Brassica_oleracea_var._italica_HQ871864
Raphanus_sativus_X78452
...
Base composition:
a c g t
0.239 0.240 0.258 0.263
Phylogenetische Rekonstruktion
Um einen einfachen Neighbour-joining-Baum aus dem Alignment zu erstellen, berechnet man zuerst eine Distanzmatrix, dann den Baum.
# Distanzmatrix
> dst <- dist.dna(al2)
# NJ-Baum
> tre <- nj(dst)
> tre
Phylogenetic tree with 7 tips and 5 internal nodes.
Tip labels:
Brassica_rapa_subsp._pekinensis_AB901369, Brassica_rapa_subsp._...
Unrooted; includes branch lengths.
> plot(tre)
Phylogenetische Rekonstruktion

Maximum Parsimony
Maximum Parsimony beruht auf der Annahme einer "sparsamen" Evolution.
> library(phangorn)
> library(ape)
> phy <- phyDat(al2) # Daten umwandeln zu phyDat-Format
> phy
7 sequences with 1159 character and 78 different site patterns.
The states are a c g t
# Suche einen maximal parsimonischen Baum fuer phy
# starte mit tre
> mp <- optim.parsimony(tre, phy)
Final p-score 454 after 0 nni operations
> mp
Phylogenetic tree with 7 tips and 5 internal nodes.
Tip labels:
Brassica_rapa_subsp._pekinensis_AB901369, Brassica_rap ...
Unrooted; no branch lengths.
Maximum Parsimony
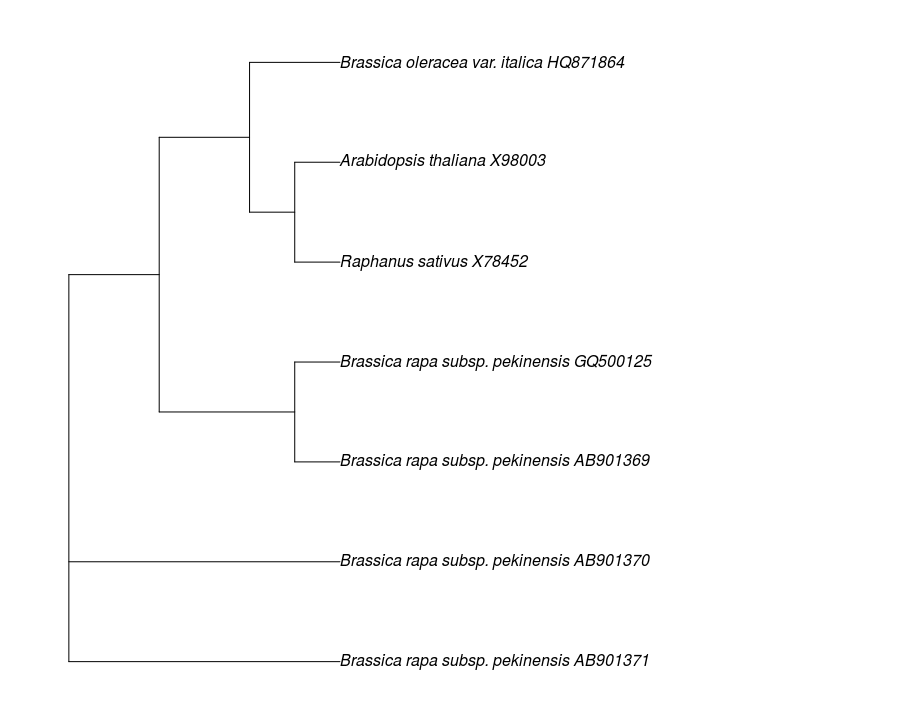
Maximum Likelihood
Maximum Likelihood ist das methodisch aufwändigste, wenn auch zuverlässigste Rekonstruktionsverfahren. Es ist ebenso über das Paket "phangorn" zugänglich, wird hier aber nicht weiter behandelt.
GBIF
GBIF
- GBIF ist eine Online-Biodiversitätsdatenbank
- Realisiert als Metadatenbank
- Daten kommen von externen Teilnehmern
- Unterschiedlichste Datenbanken nehmen teil
- Über "Connector"-Software können fremde Konzepte und Strukturen an GBIF angepasst werden
GBIF
GBIF liefert "Occurrence-Data":
Welches Objekt ist wann wo angetroffen worden?
Welches Sammlungsobjekt ist wo gelagert und wo und wann wurde es gesammelt?
Objekte sind vorwiegend Organismen jeder Art. Es können Rückschlüsse über die geographische und temporale Verteilung von Biodiversität gezogen werden.
GBIF
Der programmatische Zugriff auf das Datenreservoir von GBIF ist über das dismo-Paket möglich.
Die Funktion gbif() lädt occurrence data nach bestimmten Suchbegriffen herunter und organisiert sie in einem Dataframe.
# Pakete installieren und laden
> install.packages("dismo")
> library(dismo)
# Daten von GBIF holen
> ara <- gbif("Arabis", "blepharophylla", geo = T)
531 records found
0-300-531 records downloaded
> str(ara)
'data.frame': 531 obs. of 126 variables:
$ acceptedScientificName : chr "Arabis bleph...
GIS in R
GIS in R
Es gibt zahlreiche Pakete, die es erlauben Geoinformationen mit R zu bearbeiten und zu visualisieren.
- Das Datenpaket wrld_simple enthält eine georeferenzierte Weltkarte.
- Das Paket googleVis erlaubt die Interaktion mit Google Maps.
- Das Paket maps erlaubt es, Karten zu zeichnen
- …
Eine einfache Karte
… ist schnell geplottet.
> library(maps)
> map('world')
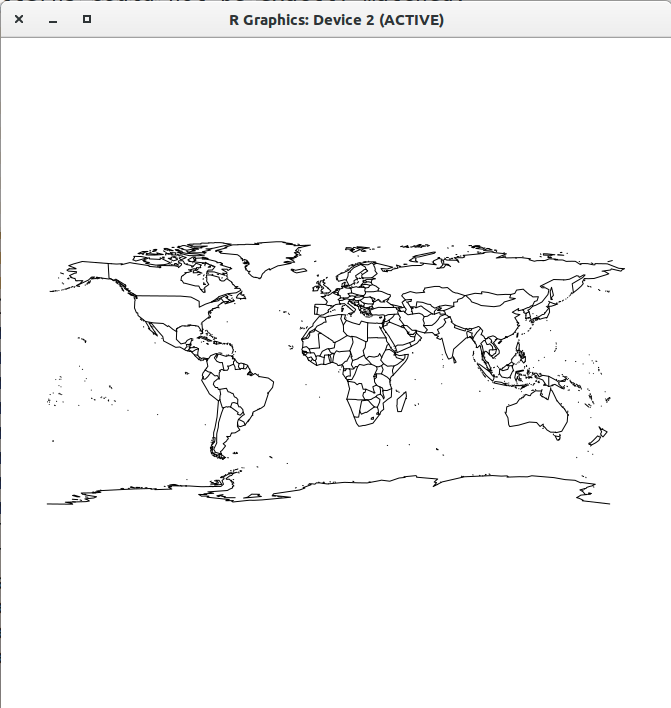
> map('world',
regions=c('Germany',
'Austria'))

> map('world', plot=F, namesonly=T)
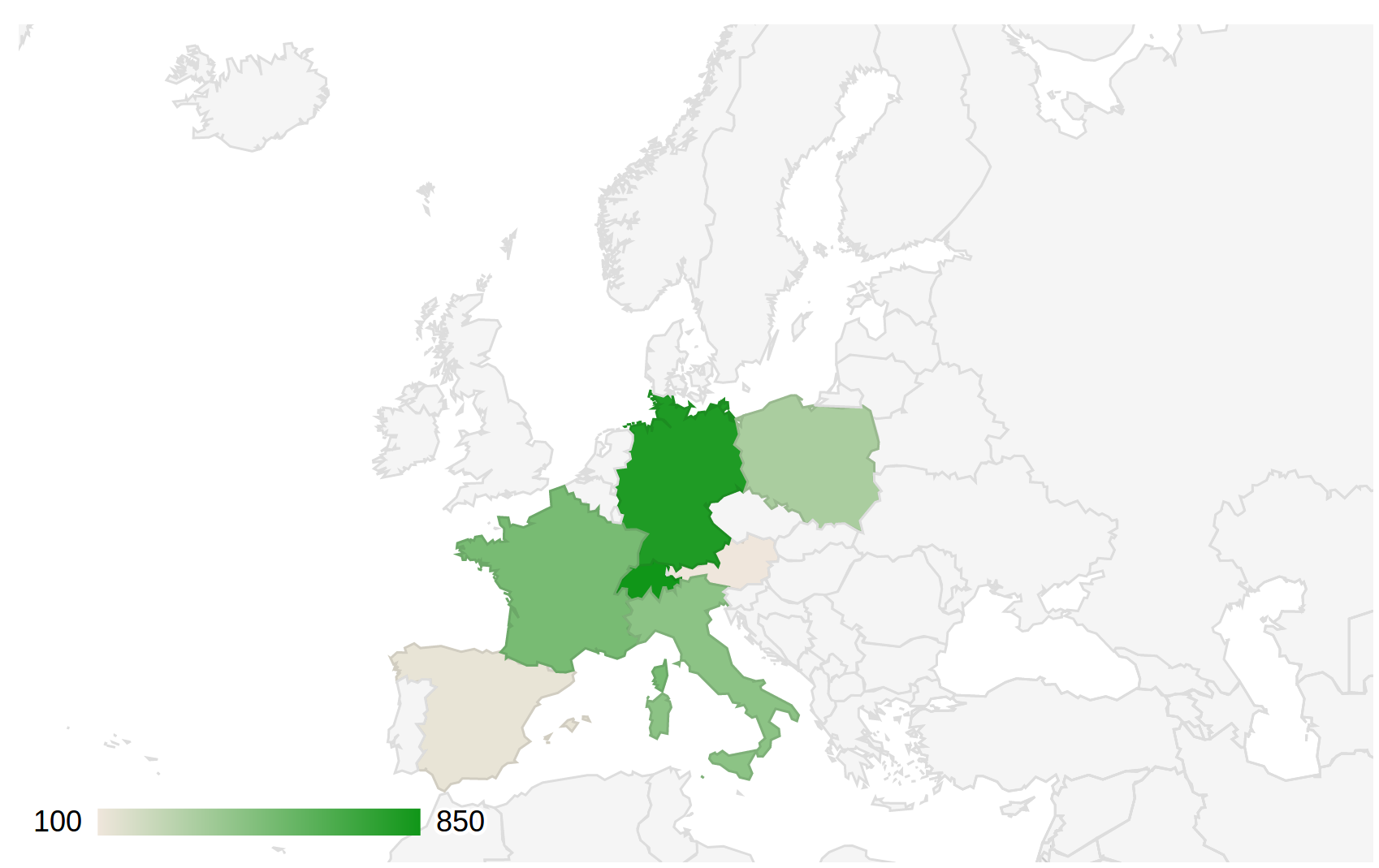
Eine Heatmap
# Pakete installieren und laden
> install.packages("googleVis")
> library(googleVis)
# Daten vorbereiten
> land <- c('Germany','Austria','France','Switzerland','Italy','Spain','Poland')
> bdidx <- c(800,100,500,850,432,123,333)
> inp <- data.frame(land, bdidx)
> inp
land bdidx
1 Germany 800
2 Austria 100
3 France 500
4 Switzerland 850
5 Italy 432
6 Spain 123
7 Poland 333
geo<-gvisGeoChart(inp,locationvar='land',colorvar='bdidx',options=list(region="150"))
plot(Geo)
Eine Heatmap
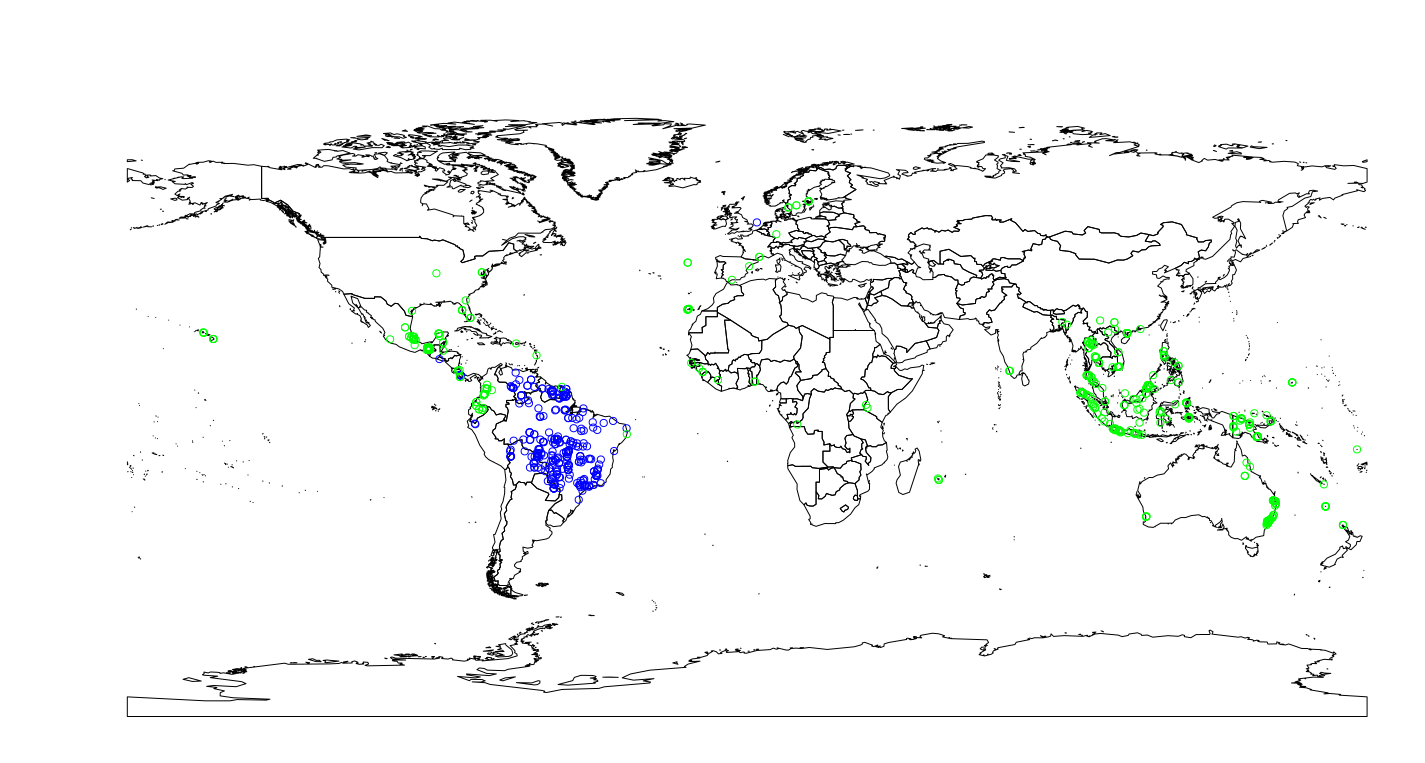
Verbreitungsdaten
> library(dismo)
> library(maps)
# Daten von GBIF holen
mb <- gbif("Musa", "acuminata", geo = T)
aa <- gbif("Ananas", "ananassoides", geo = T)
data(wrld_simpl)
plot(wrld_simpl)
points(mb$lon, mb$lat, col='red')
points(aa$lon, aa$lat, col='blue')
Obstverbreitung