Stringr
- Stringr ist ein Paket, das weitere Funktionen zur Stringbearbeitung zur Verfügung stellt.
- Seine Syntax ist außerdem konsistenter als bei vergleichbaren "eingebauten" Funktionen.
(Stringr:) Regular Expressions
- .: ein beliebiges Zeichen
- [a-z] ein Zeichen aus einer bestimmten Zeichenmenge
- .+: mindestens ein oder mehr Zeichen
- .*: kein, ein oder mehrere Zeichen
- [atgc]{2,4} zwei bis vier DNA-Basen
Welche(s) Muster findet atg[tc]{3,9}.*a+?
Stringr – GC-Gehalt
Z.B.: Die Stringr-Funktionen str_count() und str_length().
# stringr laden
> library(stringr)
> a <- c('tgactgatctagctgact','atgctactgtagctagc','gatcagctagcttctcgtct')
# Wie oft kommt g oder c in den Sequenzen vor?
> str_count(a,'[gc]')
[1] 8 8 10
# Wie lang sind die Sequenzen?
> str_length(a)
[1] 18 17 20
# Prozentualen GC-Gehalt berechnen:
> str_count(a,'[gc]')/str_length(a)*100
[1] 44.44444 47.05882 50.00000
Welche Funktionen enthält…
…Stringr noch?
# Wir benutzen die library-Funktion mit dem help-Parameter:
> library(help='stringr')
Tidyverse
- Die beiden libraries tidyr und dplyr sind nützlich zur Umformung von Daten
- Oft müssen Daten für die Analyse in einer bestimmten Form vorliegen.
- Ebenso oft steht man vor dem Problem, dass Rohdaten nicht auf diese Art gespeichert vorliegen.
tidyr – gather()
> strain <- c(1,2,3)
> adh_str <- rnorm(3, mean=10)
> apx_str <- rnorm(3, mean=11)
> adh_kon <- rnorm(3, mean=5)
> apx_kon <- rnorm(3, mean=4)
> d <- data.frame(strain,adh_kon,adh_str,apx_kon,apx_str)
> d
strain adh_kon adh_str apx_kon apx_str
1 1 6.382343 9.655073 4.026705 11.39814
2 2 4.878100 9.026552 6.165706 10.79602
3 3 5.127867 9.967047 3.030694 10.04184
> d %>% gather(strain, adh_kon:apx_str)
strain adh_kon:apx_str
1 1 adh_kon 6.382343
2 2 adh_kon 4.878100
3 3 adh_kon 5.127867
4 1 adh_str 9.655073
5 2 adh_str 9.026552
6 3 adh_str 9.967047
7 1 apx_kon 4.026705
8 2 apx_kon 6.165706
9 3 apx_kon 3.030694
10 1 apx_str 11.398141
11 2 apx_str 10.796025
12 3 apx_str 10.041840
Infix vs Prefix
Prefix-Funktion:
func(arg1, arg2)
Infix-Funktion:
arg1 fun arg2
# zB.
a * b # infix
`*`(a, b) # prefix
Pipe-Operator
Der Infix-Pipe-Operator ist Teil des Paketes dplyr.
> function1() %>% function2()
Arbeitet wie eine Leitungsverbindung ("Pipe") von der Ausgabe einer Funktion zur Eingabe einer anderen.
# Was macht
> `:`(10,1) %>% mean #?
dplyr – distinct()
> d <- as.integer(rnorm(1000, mean=3))
> d <- data.frame(d)
> d
1 3
...
998 2
999 3
1000 2
> e <- d %>% distinct() # oder distinct(d)
> e
d
1 1
2 2
3 3
4 4
5 0
6 5
7 6
# als Test: Wie bildet man den Durchschnitt aus e?
dplyr – filter()
Welche Iris-spezies hat die längsten Sepalen?
> filter(iris, Sepal.Length > 7) %>% select(Species, Sepal.Length)
Species Sepal.Length
1 virginica 7.1
2 virginica 7.6
3 virginica 7.3
4 virginica 7.2
5 virginica 7.7
6 virginica 7.7
7 virginica 7.7
8 virginica 7.2
9 virginica 7.2
10 virginica 7.4
11 virginica 7.9
12 virginica 7.7
> filter(iris, Sepal.Length > 7) %>% select(Species) %>% distinct()
Species
1 virginica
dplyr – sampling
Iris-Lotterie…
# ich haett gern 3 beliebige Iris-Zeilen
> sample_n(iris, 3)
Sepal.Length Sepal.Width Petal.Length Petal.Width Species
39 4.4 3.0 1.3 0.2 setosa
6 5.4 3.9 1.7 0.4 setosa
74 6.1 2.8 4.7 1.2 versicolor
# ...und die (zufaellige) Haelfte der setosas
> filter(iris, Species == "setosa") %>% sample_frac(iris, 0.5)
Sepal.Length Sepal.Width Petal.Length Petal.Width Species
47 5.1 3.8 1.6 0.2 setosa
40 5.1 3.4 1.5 0.2 setosa
12 4.8 3.4 1.6 0.2 setosa
32 5.4 3.4 1.5 0.4 setosa
9 4.4 2.9 1.4 0.2 setosa
16 5.7 4.4 1.5 0.4 setosa
...
dplyr – summarise()
Was sind die Eckdaten der Iris-Sepalen?
> summarise(iris, avg=mean(Sepal.Length),
mn=min(Sepal.Length),mx=max(Sepal.Length))
avg minim maxim
1 5.843333 4.3 7.9
dplyr – bind_rows()
Wie verbindet man unterschiedliche Dataframes?
> bind_rows(iris[1:3,],mtcars[1:3,])
Sepal.Length Sepal.Width Petal.Length Petal.Width Species mpg cyl disp hp drat wt qsec vs am gear carb
1 5.1 3.5 1.4 0.2 setosa NA NA NA NA NA NA NA NA NA NA NA
2 4.9 3.0 1.4 0.2 setosa NA NA NA NA NA NA NA NA NA NA NA
3 4.7 3.2 1.3 0.2 setosa NA NA NA NA NA NA NA NA NA NA NA
4 NA NA NA NA NA 21.0 6 160 110 3.90 2.620 16.46 0 1 4 4
5 NA NA NA NA NA 21.0 6 160 110 3.90 2.875 17.02 0 1 4 4
6 NA NA NA NA NA 22.8 4 108 93 3.85 2.320 18.61 1 1 4 1
dplyr – intersect()
Schnittmenge zwischen Dataframes:
> d
a b
1 A 1
2 B 2
3 C 3
> e
a b
1 D 1
2 E 2
3 C 3
> intersect(d,e)
a b
1 C 3
apply – dplyr-Style
Nicht zuletzt bietet dplyr eine Alternative zu den etwas unübersichtlichen apply-Funktionen des Basis-R.
ldply() nimmt z.B. eine Liste an und gibt einen Dataframe aus.
Plotly
- Plotly ist eine extrem mächtige R-Funktionssammlung für Diagramme und Grafiken.
- Es ermöglicht Animationen, 3D-Grafik und Online-Anbindung.
- Es hat zahlreiche Abhängigkeiten zu anderen Paketen.
Einfacher Balkenplot
> install.packages{'plotly'}
> library{plotly}
> tax <- c('A.thaliana','A.lyrata','A.petraea')
> individuals <- c(12,14,23)
> p<-plot_ly(x = tax, y = individuals, name='Arabidopsis samples', type='bar')
Einfacher Balkenplot
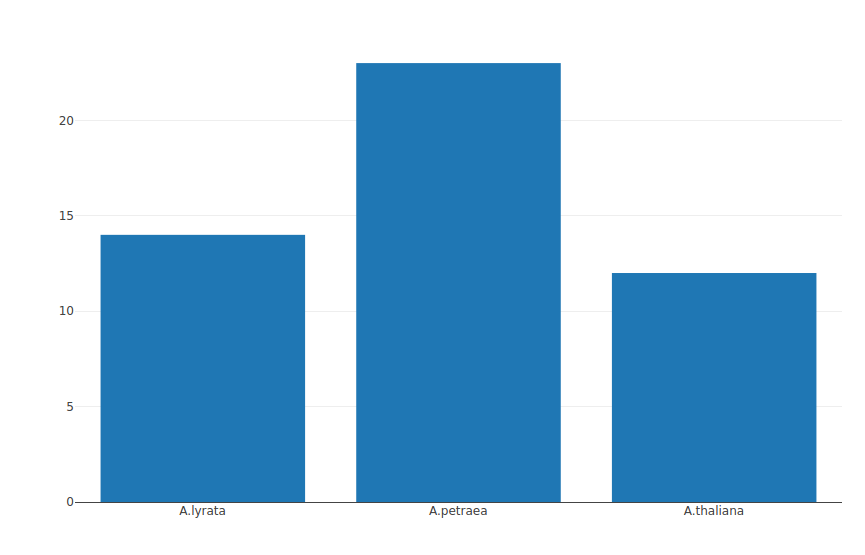
Gruppierter Balkenplot
> tax <- c('A.thaliana','A.lyrata','A.petraea')
> ind_au <- c(234,422,129) # in Österreich
> ind_de <- c(334,222,228) # in Deutschland
> ind_su <- c(434,162,189) # in der Schweiz
> ara <- data.frame(tax, ind_de, ind_au, ind_su)
> p <- plot_ly(ara, x=~tax,y=~ind_de,name="Deu", type='bar') %>%
add_trace(y=~ind_au,name='Aus', type='bar') %>%
add_trace(y=~ind_su,name='Sui', type='bar')
> p
Gruppierter Balkenplot
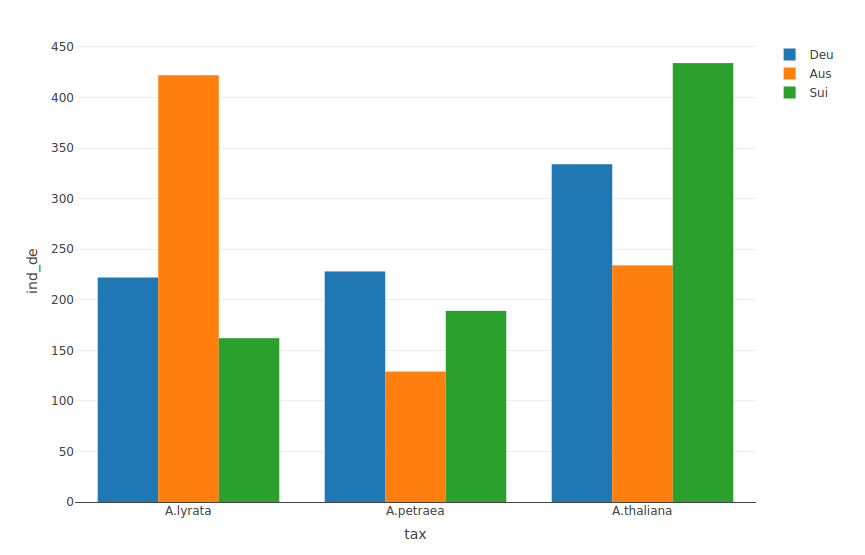
Gestapelter Balkenplot
> p <- plot_ly(ara, x=~tax,y=~ind_de,name="Deu", type='bar') %>%
add_trace(y=~ind_au,name='Aus', type='bar') %>%
add_trace(y=~ind_su,name='Sui', type='bar') %>%
layout(yaxis = list(title = "Individuals"), barmode = 'stack')
> p
Gestapelter Balkenplot

Beschriftung
Folgendes erzeugt eine Beschriftung in den Balken(teilen). Es muß in der Definition der betreffenden Traces eingefügt werden.
... ,text = daten, textposition = 'auto' ...
Was macht…?
...xaxis = list(title="", tickangle=55), margin = list(b = 100)...
Farben
col1 <- list(color = 'rgb(255,100,100)')
col2 <- list(color = 'rgb(100,255,100)')
col3 <- list(color = 'rgb(100,100,255)')
p <- plot_ly(ara, x=~tax,y=~ind_de,name="Deu", type='bar',
text=ind_de,textposition='auto', marker=col1) %>%
add_trace(y=~ind_au,name='Aus', type='bar',text = ind_au,textposition='auto',
marker=col2) %>%
add_trace(y=~ind_su,name='Sui', type='bar',text = ind_su,textposition='auto',
marker=col3) %>%
layout(yaxis = list(title = "Individuals"), xaxis = list(title="",
tickangle=55), barmode = 'group',margin= list(b=110))
Lineplot
> x <- 1:1000
> y <- rnorm(1000, mean = 100)
> p <- plot_ly(x = ~x, y = ~y, type = 'scatter', mode = 'lines')
> p
Lineplot
Lineplot
> x <- 1:100
> y <- rnorm(100, mean = 10)
> p <- plot_ly(x = ~x, y = ~y, type = 'scatter', mode = 'lines+markers')
> p
Lineplot
...mode = 'markers'...?
Mehr Lineplot
> x <- 1:1000
> x2 <- 1:100
> y <- rnorm(1000, mean = 100)
> y2 <-rnorm(100,mean=-5)
> p <- plot_ly(x = ~x, y = ~y, type = 'scatter', mode = 'lines') %>%
add_trace(x = ~x2, y = ~y2, type = 'scatter', mode = 'lines')
> p
Mehr Lineplot
Noch mehr Lineplot
> p <- plot_ly(x = ~x, y = ~y1, type = 'scatter', mode = 'lines') %>%
add_trace(x = ~x, y = ~y2, type = 'scatter', mode = 'lines',
line = list(dash = 'dot')) %>%
add_trace(x = ~x, y = ~y3, type = 'scatter', mode = 'lines',
line = list(dash = 'dash', color = 'rgb(255,0,0)'))
Noch mehr Lineplot
 Gemischter Bar- und Lineplot?
Gemischter Bar- und Lineplot?
Lücken
> x <- 1:8
> y1 <- c(2, 4, NA, 2, 3, NA, 5, 2)
> y2 <- y1 + 1
> p <- plot_ly(x = ~x, y = ~y1, type = 'scatter', mode = 'lines') %>%
add_trace(x = ~x, y = ~y2, type = 'scatter', mode = 'lines',
line = list(dash = 'dash'), connectgaps = T)
Lücken
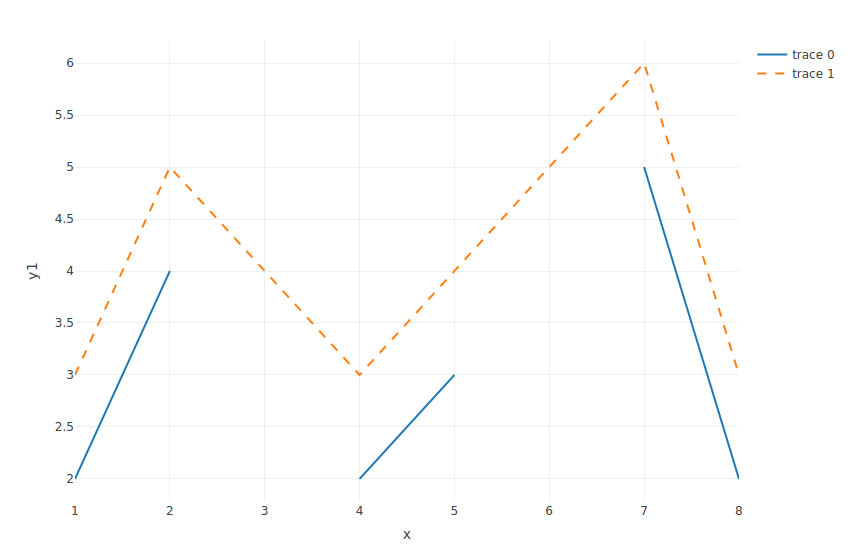 NA bedeuted "fehlende Daten".
NA bedeuted "fehlende Daten".
Interpolation
Unsere Daten sind einzelne Datenpunkte. Die Zwischenräume können auf unterschiedliche Art interpoliert werden. (Sofern das sinnvoll ist…)
> x <- 1:8
> y1 <- c(2, 4, 5, 2, 3, 7, 5, 2)
> y2 <- y1 + 5
> y3 <- y1 + 10
> y4 <- y1 + 15
> p <- plot_ly(x = ~x) %>%
add_lines(y = ~y1, , name = "linear", line = list(shape = "linear")) %>%
add_lines(y = ~y2, , name = "spline", line = list(shape = "spline")) %>%
add_lines(y = ~y3, , name = "hvh", line = list(shape = "hvh")) %>%
add_lines(y = ~y4, , name = "vhv", line = list(shape = "vhv"))
Interpolation
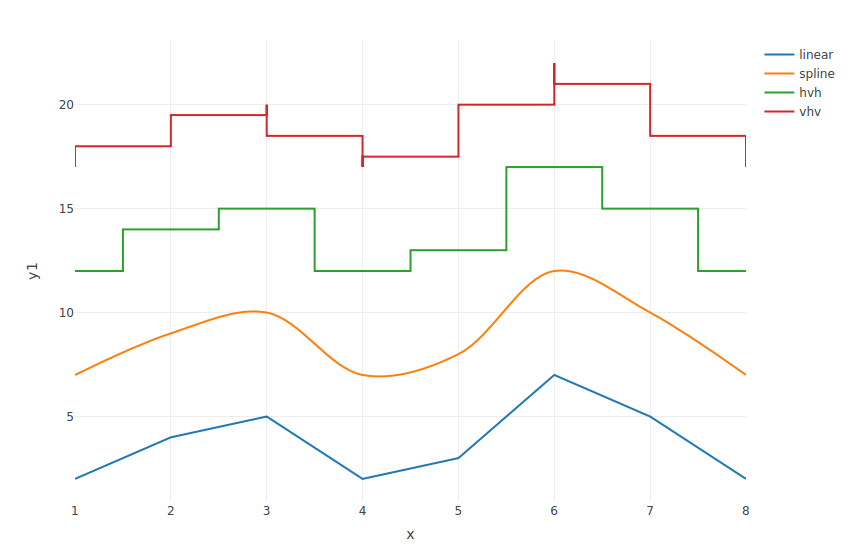
Iris reloaded
> p <- plot_ly(data = iris, x = ~Sepal.Length, y = ~Petal.Length, type="scatter",mode="markers",
+ marker = list(size = 10,
+ color = 'rgba(240, 55, 44, .2)',
+ line = list(color = 'rgba(50, 215, 50, 1)',
+ width = 2))) %>%
+ layout(title = 'Iris schon wieder',
+ yaxis = list(zeroline = FALSE),
+ xaxis = list(zeroline = FALSE))
> p
Iris reloaded

Was macht…
...symbol = ~Species, symbols = c('circle','x','o')...?
Hypothesentests
Allgemeines Vorgehen:
- Man stellt zu einer Fragestellung zwei Hypothesen auf: Nullhypothese H0 und Alternativhypothese H1.
- Die Hypothesen sind so formuliert, dass man die Nullhypthese widerlegen möchte, z.B. "Die Mittelwerte zweier Proben unterscheiden sich nicht signifikant."
- Ein Signifikanzniveau a muss festgelegt werden. Häufig wird a = 5 % gewählt.
Hypothesentests
Statistische Fehlentscheidungen
- Fehler 1. Art oder α-Fehler: Nullhypothese wird fälschlicherweise abgelehnt, obwohl sie wahr ist.
- Fehler 2. Art oder β-Fehler: Nullhypothese wird fäschlicherweise angenommen, obwohl sie falsch ist.
Vergleich zweier Proben
Auswahl häufig genutzter statistischer Methoden:
- Vergleich zweier Varianzen: Fishers F-test (var.test())
- Vergleich zweier Mittelwerte (setzt Normalverteilung und gleiche Varianz voraus): t-Test (t.test())
- Test der Unabhängigkeit in Kontingenztafeln: Chi-Square Test (chisq.test()) oder exakter Fishers Test (fisher.test())
Fishers F-test
Als Beispiel generieren wir einen Datensatz der Anzahl gut entwickelter Samen pro Schote zweier unterschiedlicher Boechera divaricarpa Pflanzen:
# Zunächst lesen wir den Datensatz ein. Ist die Datei nicht im
# Arbeitsverzeichnis muss der Pfadname zur Datei angegeben sein.
> samen <- read.table("Samen.txt", header = T)
> attach(samen) # ...to search path
> names(samen)
# Nun koennen die Varianzen ausgelesen werden
> var(Pflanze1)
[1] 68.44444
> var(Pflanze2)
[1] 84.88889
# Nun teilen wir die groessere durch die kleinere Varianz
> ratio <- var(Pflanze2)/var(Pflanze1)
> ratio
[1] 1.24026
Fishers F-test
Die Null Hypothese ist, dass beide Varianzen nicht signifikant unterschiedlich sind. Wir lesen den Signifikanzwert p aus der F-Verteilung (Fisher Verteilung).
# 9 und 9 sind die Freiheitsgrade
> 2*(1-pf(ratio, 9, 9))
[1] 0.7536297
Die Null-Hypothese hat Bestand.
Fishers F-test
Einfacher geht es mit:
> var.test(Pflanze1, Pflanze2)
F test to compare two variances
data: Pflanze1 and Pflanze2
F = 0.80628, num df = 9, denom df = 9, p-value = 0.7536
alternative hypothesis: true ratio of variances is not equal to 1
95 percent confidence interval:
0.2002692 3.2460895
sample estimates:
ratio of variances
0.8062827
Diese Daten dürfen mit einem t-Test verglichen werden!
t-Test
Fragestellung hier: Sind die Mittelwerte zweier unabhängiger Stichproben unterschiedlich? Formel für den t-Test: $$ t = \frac{\text{Mittelwert von Stichprobe 1} - \text{Mittelwert von Stichprobe 2}}{\text{Standardfehler der Differenzen beider Mittelwerte}} $$ $$ df = n1 + n2 - 2 $$ Der t-Test bezieht sich auf den Standardfehler (SEM, standard error of the mean): $$ \text{(geschätzter) Standardfehler} = \frac{s}{\sqrt{n}} = \frac{\sqrt{\frac{\sum_{t=1}^{n}(x_1-\bar{x})^2}{n-1}}}{\sqrt{n}} $$t-Test
Als Beispiel schauen wir uns an, ob die Mittelwerte der Samenbildung in Pflanze 1 und Pflanze 2 unterschiedlich sind.
Zunächst sehen wir die Daten grafisch mit einem "Box und Whiskers Plot" an:
> boxplot(samen[,1], samen[,2], data = samen, xlab = "Pflanzen",
ylab = "entwickelte Samen", col = c("blue", "red"),
names = c("Pflanze1", "Pflanze2"), main = "Boechera divaricarpa")
t-Test
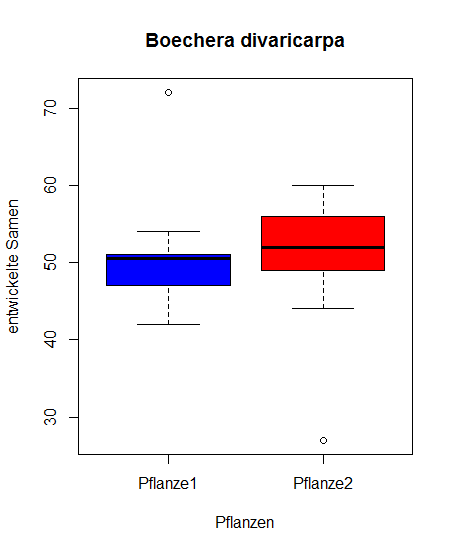
t-Test
Der t Test wird in R mit t.test() aufgerufen.Allgemeine Form:
t.test(x, ...)
t.test(x, y = NULL, alternative = c("two.sided", "less", "greater"),
mu = 0, paired = FALSE, var.equal = FALSE,
conf.level = 0.95, ...)
- x: Vektor nummerischer Daten
- y: Vektor nummerischer Daten
- mu: Differenz zwischen Mittelwerten bei Annahme der Nullhypothese
- var.equal: indiziert ob die Varianzen als gleich angenommen werden. bei TRUE werden Varianzen gepoolt, bei FALSE (default) wird die Welch-Annäherung zur Bestimmung der Freiheitsgrade genutzt
- paired: unabhängige Stichproben
t-Test
In unserem Beispiel erfolgt die Berechnung als:
# Zweiseitiger t-Test fuer zwei unabhaengige Stichproben mit gleicher Varianz
> t.test(samen[,1], samen[,2], alternative = c("two.sided"), mu = 0,
paired = FALSE, var.equal = TRUE, conf.level = 0.95)
Two Sample t-test
data: samen[, 1] and samen[, 2]
t = 0.25538, df = 18, p-value = 0.8013
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
-7.226749 9.226749
sample estimates:
mean of x mean of y
51 50
Die Nullhypothese wird hier angenommen, es liegt kein signifikanter Unterschied der Mittelwerte vor!
# Fuehrt zu demselben Resultat
> t.test(samen[,1], samen[,2], var.equal = TRUE)
t-Test
Ein weiteres Beispiel: Zwei R-Kurse mit jeweils 10 Studierenden werden mit Punkten bewertet. Vektor x sei die Punkteverteilung von Kurs a, Vektor y die Punkteverteilung von Kurs b.
> x <- c(45, 57, 42, 85, 47, 90, 75, 82, 93, 50)
> y <- c(85, 88, 77, 95, 99, 80, 74, 93, 82, 94)
t-Test
# Test der Varianzen
> var.test(x, y)
F test to compare two variances
data: x and y
F = 5.8028, num df = 9, denom df = 9, p-value = 0.01518
alternative hypothesis: true ratio of variances is not equal to 1
95 percent confidence interval:
1.441344 23.362213
sample estimates:
ratio of variances
5.802843
# Box und Whiskers Plot
> boxplot(x, y, ylab = "Punkte", names = c("Kurs 1", "Kurs 2"),
col = c("orange", "red"), main = "Kursbewertungen")
t-Test
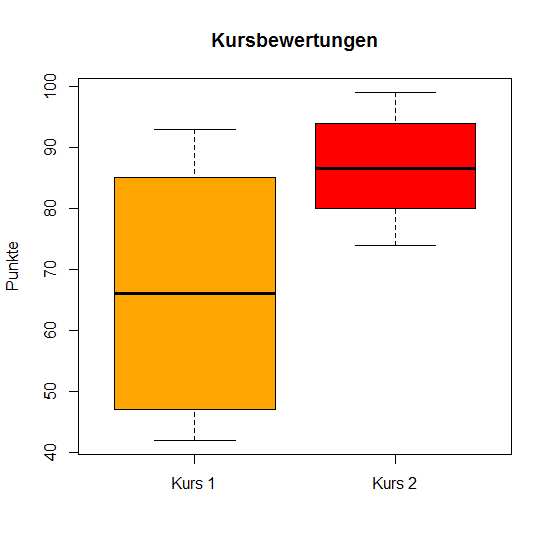
Welch t-Test
Welchs t-Test geht von zwei normalverteilten Populationen mit ungleicher Varianz und der Hypothese aus, daß diese gleiche Mittelwerte haben.
# Nun ein Welch t-Test mit Standard Parametern
> t.test(x, y)
Welch Two Sample t-test
data: x and y
t = -2.8897, df = 12.012, p-value = 0.01357
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
-35.25367 -4.94633
sample estimates:
mean of x mean of y
66.6 86.7
Wilcoxon Rank Test
- nicht parametrischer Test (basiert nicht auf Verteilungen)
- kann benutzt werden, wenn keine Normalverteilung gegeben ist
- hier nicht weiter besprochen
Chi-Square Test
Dient dem Vergleich zweier Häufigkeiten bzw Verteilungen.
$$ \chi^2 = \sum\frac{(\text{beobachtete~Häufigkeit} - \text{erwartete~Häufigkeit})^2}{\text{erwartete~Häufigkeit}} $$Chi-Square Test
Der Chi-Square Test wird häufig im Kontext der Genetik eingesetzt.
Als Beispiel fragen wir, ob in Mutanten Arabidopsis-Linien eine Antibiotikum-Resistenz nach Mendel segregiert.
Chi-Square Test
In einer Arabidopsis-Linie haben wir 263 resistente Keimlinge und 93 sensitive Keimlinge ausgezählt. Das erwartete Verhältnis nach Mendel ist 3:1 resistent:sensitiv.
# Generieren der Matrix
> colnames = c("beobachtet", "erwartet")
> rownames = c("resistent", "sensitiv")
> genetik <- matrix(c(263, 93, 267, 89), byrow = FALSE,
nrow = 2, dimnames = list(rownames, colnames))
> genetik
beobachtet erwartet
resistent 263 267
sensitiv 93 89
> chi <- ((263-267)^2/267)+((93-89)^2/89)
> chi
[1] 0.2397004
Chi-Square Test
Um den kritischen Wert für die Chi-Square Verteilung nachzusehen, benötigen wir die Freiheitsgrade (hier: 1) und das α-Niveau (hier: p = 0.95).
# Auslesen des kritischen Wertes
> qchisq(0.95, 1)
[1] 3.841459
Wichtig: Ist der statistische Wert größer als der kritische Wert, wird die Null-Hypothese abgelehnt!
Chi-Square Test
# Jetzt lassen wir R rechnen...
> count <- matrix(c(263, 93), nrow = 2)
> count
[,1]
[1,] 263
[2,] 93
> chisq.test(count, p = c(3/4, 1/4))
Chi-squared test for given probabilities
data: count
X-squared = 0.2397, df = 1, p-value = 0.6244
Exakter Fisher Test
- Signifikanztest auf Unabhängigkeit in Kontingenztafeln
- auch für kleinen Stichprobenumfang
- hier nicht weiter besprochen
Lineare Regression
- statistisches Verfahren, um eine beobachtete abhängige Variable durch eine oder mehrere unabhängige Variablen zu beschreiben
- es liegen zwei metrische Größen zugrunde: eine Einflussgröße x und eine Zielgröße y.
- y als Zielgröße
- x als Einflussgröße
- a, b als Koeffizienten
Die generelle mathematische Form ist: y = ax + b
Lineare Regression
Für die Regression rufen wir in R die Funktion lm() auf.
Als Beispiel vergleichen wir eine Funktion von 10 Individuen.
# Erstellen der Vektoren fuer Groesse und Gewicht der Personen
> groesse <- c(143, 152, 163, 198, 201, 174, 178, 169, 183, 192)
> gewicht <- c(51, 63, 59, 110, 87, 89, 70, 72, 85, 99)
# Veranschaulichung
> plot(gewicht, groesse, col = "blue",
main = "Groesse und Gewicht Regression",
abline(lm(groesse ~ gewicht)),cex = 1.3,
pch = 16,xlab = "Gewicht in Kg",ylab = "Groesse in cm")
Lineare Regression

Lineare Regression
# Mit lm() werden die beiden Koeffizienten (a, b) ausgegeben
> lm(gewicht ~ groesse)
Call:
lm(formula = gewicht ~ groesse)
Coefficients:
(Intercept) groesse
-71.2707 0.8544
Ziel ist es, die Reste, bzw die Quadratwerte der Reste zu minimieren.
PCA
Pricipal component analysis
Eine Technik, um vieldimensionale Datensätze unter Beibehaltung von möglichst viel Information so weit an Dimensionalität zu reduzieren, daß sie leichter auswert- und visualisierbar sind.
PCA
Ein Datensatz mit n Messungen von p Merkmalen wird typischerweise als Matrix dargestellt.
Bei zahlreichen Messungen/Merkmalen ist es schwer, zu erkennen, welche Messwerte Trends aufweisen oder überhaupt relevant für die betreffenden Proben sind.
PCA
Man kann einen solchen Datensatz auch als Liste von Punktkoordinaten oder Vektoren in einem p-dimensionalen Raum interpretieren.
Mehr als 3 Dimensionen sind allerdings schwer zu veranschaulichen…
PCA
Man versucht deshalb, diese Punkte mit minimalem Informationsverlust in einen max. dreidimensionalen Unterraum zu projizieren.Voraussetzung ist weitgehende Normalverteilung des Datensatzes.
PCA - Anwendungen
Beispiel
- Messung zahlreicher morphologischer Merkmale aus Individuen einer Hybridpopulation (Blattlängen, -breiten, Internodienlänge, -dicke, -zahl, Blatthaardichte, Verzweigungswinkel…)
- Welche Individuen sind ähnlicher zu welcher Elternart? Gibt es überhaupt eine Differenzierung? Welche Merkmale sind besonders aussagekräftig?
PCA - Vorgehen
- Die erste Hauptkomponente (PC1) wird so gesucht, daß man eine Linie durch die die Daten repräsentierende Punktewolke sucht, die die Quadrate der Abweichungen der Punkte (Fehler) minimiert.
- Die zweite Hauptkomponente (PC2) steht orthogonal auf PC1 und minimiert wiederum die Fehlerquadrate.
- Die folgenden Hauptkomponenten stehen jeweils orthogonal auf allen anderen usw.
PCA - Vorgehen
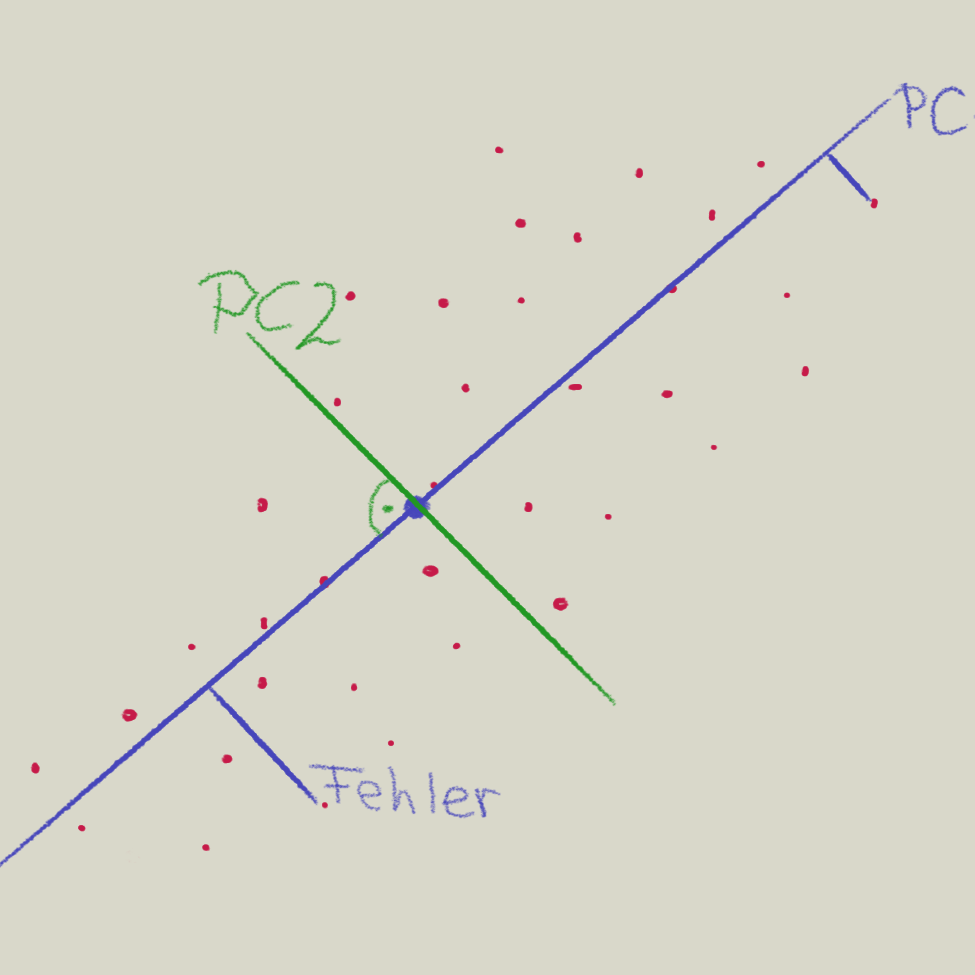
Wieviele und welche Hauptkomponenten für die weitergehende Analyse der Daten am geeignetsten sind ist abhängig von den Daten.
PCA in R
Es gibt mehrere R-Pakete und Funktionen, die die Durchführung einer PCA erlauben.
Die nachfolgende Visualisierung und weitere Analyse kann wiederum auf unterschiedliche Art erfolgen und auch hier gibt es viele Möglichkeiten in R.
Die im Folgenden beschriebene Herangehensweise ist also nur als Beispiel zu verstehen.
A propos Beispiel: wir verwenden wiederum…den Iris-Datensatz.
PCA in R
Wie sah der iris-Datensatz nochmal aus?
> data(iris)
> head(iris, 5)
Sepal.Length Sepal.Width Petal.Length Petal.Width Species
1 5.1 3.5 1.4 0.2 setosa
2 4.9 3.0 1.4 0.2 setosa
3 4.7 3.2 1.3 0.2 setosa
4 4.6 3.1 1.5 0.2 setosa
5 5.0 3.6 1.4 0.2 setosa
...
PCA in R
Zuerst laden/installieren wir die benötigten Pakete und ihre Abhängigkeiten.
# devtools stellt eine Funktion bereit, um aus dem Software-repo
# 'Github' zu installieren.
> install.packages('devtools') # if necessary
> library(devtools)
# Downloade und erstelle Paket 'ggbiplot' von Github-User 'vqv':
> install_github("vqv/ggbiplot") # if necessary
> library(ggbiplot)
PCA in R
PCA vorbereiten und rechnen:
# Datenspalten von iris in Log-Skala umwandeln
> irlg <- log(iris[, 1:4])
# Artnamen extrahieren
> irsp <- iris[, 5]
# PCA berechnen
> irpca <- prcomp(irlg, center=T, scale.=T)
# irpca ist ein R-Objekt, das eigene Methoden zur Verfuegung stellt
# (print, predict...)
> print(irpca)
Standard deviations:
[1] 1.7124583 0.9523797 0.3647029 0.1656840
Rotation:
PC1 PC2 PC3 PC4
Sepal.Length 0.5038236 -0.45499872 0.7088547 0.19147575
Sepal.Width -0.3023682 -0.88914419 -0.3311628 -0.09125405
...
# Und barplotten:
plot(irpca, type = "b")
PCA in R
Hauptkomponenten ausgeben
> summary(irpca)
Importance of components:
PC1 PC2 PC3 PC4
Standard deviation 1.7125 0.9524 0.36470 0.16568
Proportion of Variance 0.7331 0.2268 0.03325 0.00686
Cumulative Proportion 0.7331 0.9599 0.99314 1.00000
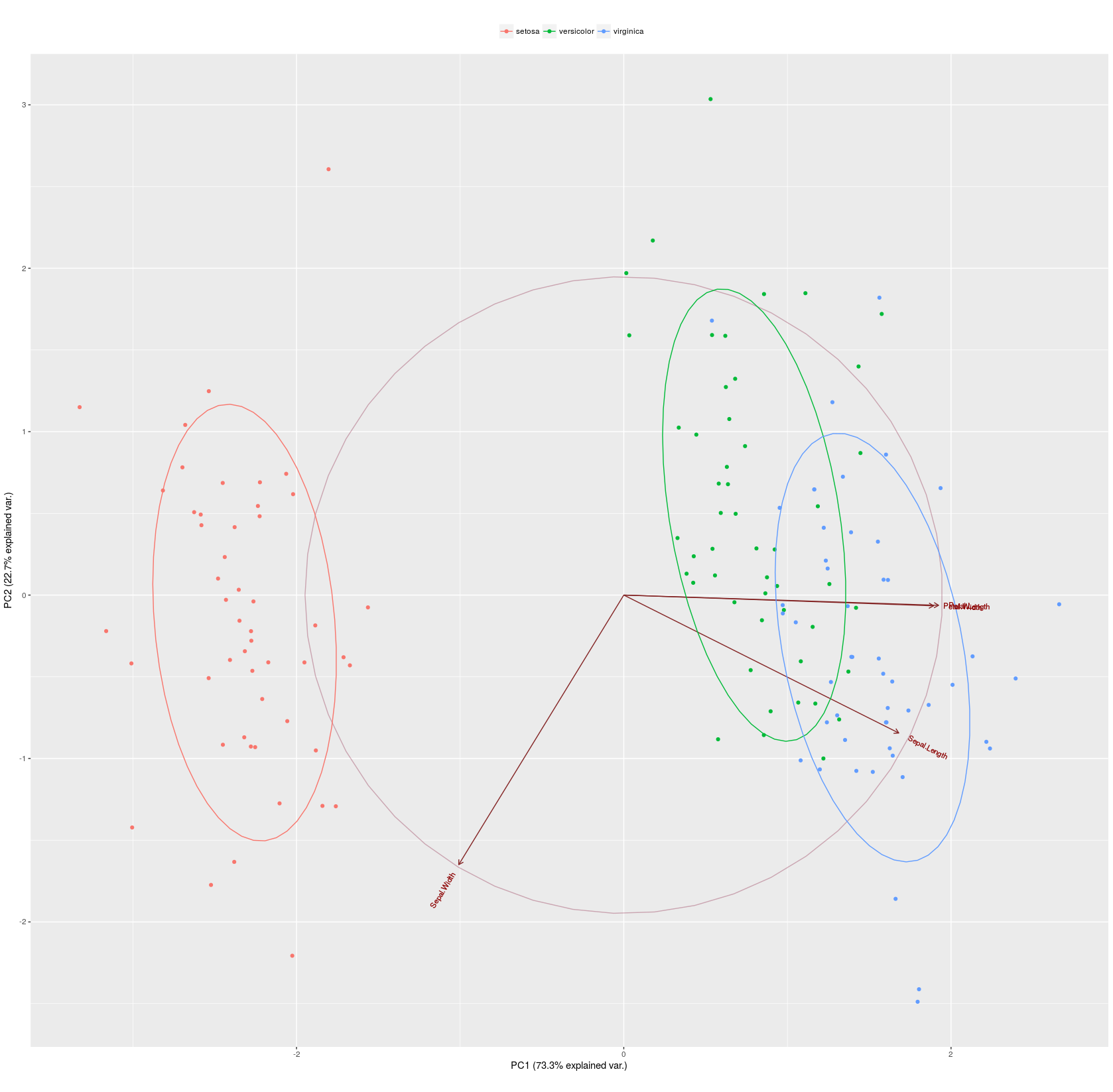
PC1 und PC2 erklären zusammen 96\% der Variabilität der Daten.
PCA in R
Zusätzliche Messungen lassen sich mit predict() passend zur vorhandenen PCA berechnen.
> Sepal.Length<-6.1
> Sepal.Width<-4.8
> Petal.Length<-2.1
> Petal.Width<-1.7
> newir<-data.frame(Sepal.Length,Sepal.Width,Petal.Length,Petal.Width)
> newir
Sepal.Length Sepal.Width Petal.Length Petal.Width
1 6.1 4.8 2.1 1.7
> predict(irpca, newdata=log(newir))
PC1 PC2 PC3 PC4
[1,] -0.8036107 -3.037735 -1.058046 0.7695977
PCA in R
Visualisierung der Daten.
> irvis <- ggbiplot(irpca, obs.scale = 1,
var.scale = 1,
groups = irsp, ellipse = T,
circle = T)
> irvis <- irvis + scale_color_discrete(
name = '')
> irvis <- irvis + theme(
legend.direction = 'horizontal',
legend.position = 'top')
> print(irvis)
PCA in R